Die Anforderungen an die Qualität und Sicherheit von Medizinprodukten werden zunehmend verschärft. Deshalb wird auch die Reinheit von Medizinprodukten immer genauer unter die Lupe genommen. In diesem Artikel erfahren Sie, wie eine zuverlässige Reinheitsbestimmung von Medizinprodukten gelingt.
Mit ihrer langjährigen Erfahrung in der chemischen Analytik und Zulassung von Medizinprodukten bietet die RMS ihren Kunden die Planung, Durchführung und Dokumentation der Reinheitsbestimmung an. Die Analysen werden nach Möglichkeit in unseren eigenen Laboratorien oder mit Einwilligung des Kunden in Zusammenarbeit mit Partnerlaboratorien durchgeführt.
Vorgehen
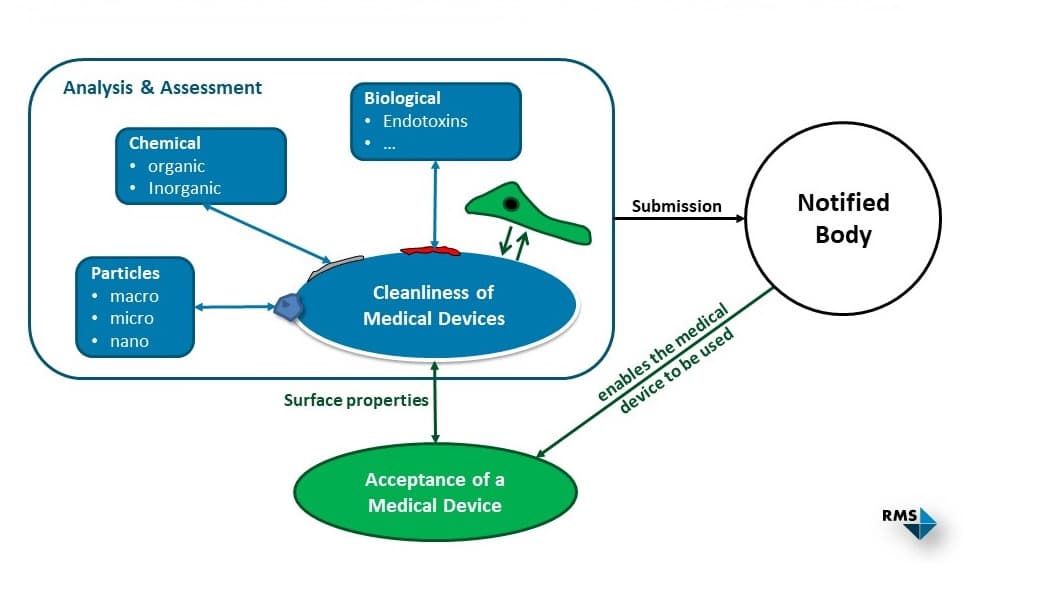
Zum Nachweis der Reinheit eines Medizinproduktes muss der Kontaminationsgrad unter vorgegebenen oder im Voraus festzulegenden Akzeptanzkriterien liegen. Die zu betrachtenden Kontaminationen können in die folgenden Klassen eingeteilt werden:
- chemische (anorganische und organische)
- biologische (z. B. Bioburden und Endotoxine)
- partikuläre Verunreinigungen
Gibt es keine produktspezifische Norm, die Akzeptanzkriterien für die Reinheit festlegt, müssen diese für die verschiedenen Kontaminationsklassen vom Hersteller selbst festgelegt und plausibel begründet werden. Dazu können z. B. verwandte Normen oder klinische Erfahrungen als Hilfestellung herangezogen werden. Gemäss ISO 19227:2018 sollte eine akzeptable Menge an Restkontaminationen weder die Funktion noch die Biokompatibilität (ISO 10993) eines Medizinprodukts beeinträchtigen.
Bei den Prüfverfahren lassen sich direkte (direkt auf Medizinprodukt messen) und indirekte (Entfernung der Verunreinigung für Prüfung) Verfahren unterscheiden. Eine direkte Analyse (z. B. XPS, FT-IR, EDX) hat den Vorteil, dass die Probenvorbereitung, welche die Ergebnisse stark beeinflussen kann, auf ein Minimum reduziert wird. Andererseits ist eine direkte und somit lokale Analyse nicht zwingend repräsentativ für das gesamte Medizinprodukt. Möchten wir aber die Reinheit des gesamten Medizinprodukts analysieren (z. B. Gesamtmenge der Partikel), wird eine Extraktion durchgeführt und der Extrakt untersucht. Organische Verunreinigungen können z. B. mittels FT-IR, HPLC, GC-MS oder TOC bestimmt werden, während anorganische Verunreinigungen mittels ICP-MS im Ultraspurenbereich analysiert werden können. Um Partikel zu klassifizieren und zu quantifizieren, müssen die Extrakte zuerst abfiltriert und der Filter optisch oder mittels REM untersucht werden. Die chemische Zusammensetzung der Partikel kann mittels FT-IR oder EDX bestimmt werden. Wie vollständig und richtig die Ergebnisse einer solchen Analyse sind, hängt stark davon ab, wie geeignet die gewählten Extraktionsbedingungen (z. B. Extraktionsmedium, Temperatur, Bewegung) für die Gewinnung der vorhandenen Kontaminationen von der Produktoberfläche sind. Die Auswahl der Extraktionsbedingungen erfolgt gemäss normativen Vorgaben (z. B. ISO 10993-12:2012) und wird spezifisch auf das zu testende Medizinprodukt abgestimmt.
Für die biologischen Untersuchungen arbeiten wir mit zuverlässigen und kompetenten Laboratorien zusammen, an die wir in Absprache mit dem Kunden Analysen weitergeben können. Falls Verunreinigungen nachgewiesen werden, müssen diese quantifiziert und ihre Bedenklichkeit muss toxikologisch bewertet werden. Dazu bieten wir literaturbasierte toxikologische Bewertungen gemäss ISO 19993-1:2018 und ISO 10993-17:2002 an.
Besprechen Sie Ihre Fragestellungen mit uns
Wir beraten Sie gerne. Ihr Kontakt für Reinheitsbestimmung ist Dr. Marc Bohner.